Chapter 1 - snakemake runs programs for you!
Bioinformatics often involves running many different programs to characterize and reduce sequencing data, and I use snakemake to help me do that.
A first, simple snakemake workflow
Here's a simple, useful snakemake workflow:
rule compare_genomes:
message: "compare all input genomes using sourmash"
shell: """
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first
sourmash compare GCF_000021665.1.fna.gz.sig \
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig \
-o compare.mat
sourmash plot compare.mat
"""
Put it in a file called Snakefile, and run it with snakemake -j 1.
This will produce the output file compare.mat.matrix.png which contains a similarity matrix and a dendrogram of the three genomes (see Figure 1).
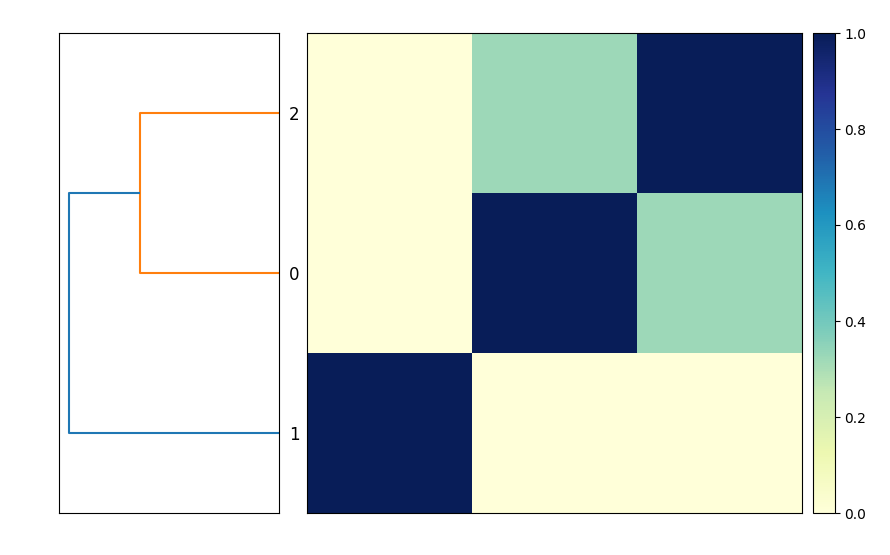
This is functionally equivalent to putting these three commands into a file compare-genomes.sh and running it with bash compare-genomes.sh -
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first
sourmash compare GCF_000021665.1.fna.gz.sig \
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig \
-o compare.mat
sourmash plot compare.mat
The snakemake version is already a little bit nicer because it will give you encouragement when the commands run successfully (with nice green text saying "1 of 1 steps (100%) done"!) and if the commands fail you'll get red text alerting you to that, too.
But! We can further improve the snakemake version over the shell script version!
Avoiding unnecessary rerunning of commands: a second snakemake workflow
The commands will run every time you invoke snakemake with snakemake -j 1. But most of the time you don't need to rerun them because you've already got the output files you wanted!
How do you get snakemake to avoid rerunning rules?
We can do that by telling snakemake what we expect the output to be by adding an output: block in front of the shell block:
rule compare_genomes:
message: "compare all input genomes using sourmash"
+ output:
+ "compare.mat.matrix.png"
shell: """
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first
and now when we run snakemake -j 1 once, it will run the commands; but when we run it again, it will say, "Nothing to be done (all requested files are present and up to date)."
The code example above looks a little odd - it's got '+' in front of a two lines, and they're colored green. What gives?
This is an example of a "diff", a line-by-line comparison of two
source code files produced by the diff program. Here, the diff
shows that we've added two lines to the original code listing - the two
lines beginning with '+'. It also adds some context above and below the
added lines so that you can more easily see where they are added to the
original code.
Below, we'll also show examples using removed lines, which will be identified with a '-' in the first position and highlighted in red.
This is because the desired output file, compare.mat.matrix.png, already exists. So snakemake knows it doesn't need to do anything!
If you remove compare.mat.matrix.png and run snakemake -j 1 again, snakemake will happily make the files again:
$ rm compare.mat.matrix.png
$ snakemake -j 1
So snakemake makes it easy to avoid re-running a set of commands if it has already produced the files you wanted. This is one of the best reasons to use a workflow system like snakemake for running bioinformatics workflows; shell scripts don't automatically avoid re-running commands.
Running only the commands you need to run
The last Snakefile above has three commands in it, but if you remove the compare.mat.matrix.png file you only need to run the last command again - the files created by the first two commands already exist and don't need to be recreated. However, snakemake doesn't know that - it treats the entire rule as one rule, and doesn't look into the shell command to work out what it doesn't need to run.
If we want to avoid re-creating the files that already exist, we need to make the Snakefile a little bit more complicated.
First, let's break out the commands into three separate rules.
rule sketch_genomes:
shell: """
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first
"""
rule compare_genomes:
shell: """
sourmash compare GCF_000021665.1.fna.gz.sig \
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig \
-o compare.mat
"""
rule plot_comparison:
message: "compare all input genomes using sourmash"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot compare.mat
"""
We didn't do anything too complicated here - we made two new rule blocks, with their own names, and split the shell commands up so that each shell command has its own rule block.
You can tell snakemake to run all three:
snakemake -j 1 sketch_genomes compare_genomes plot_comparison
and it will successfully run them all!
However, we're back to snakemake running some of the commands every time - it won't run plot_comparison every time, because compare.mat.matrix.png exists, but it will run sketch_genomes and compare_genomes repeatedly.
How do we fix this?
Adding output blocks to each rule
If add output blocks to each rule, then snakemake will only run rules where the output needs to be updated (e.g. because it doesn't exist).
Let's do that:
rule sketch_genomes:
output:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
shell: """
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first
"""
rule compare_genomes:
output:
"compare.mat"
shell: """
sourmash compare GCF_000021665.1.fna.gz.sig \
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig \
-o compare.mat
"""
rule plot_comparison:
message: "compare all input genomes using sourmash"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot compare.mat
"""
and now
snakemake -j 1 sketch_genomes compare_genomes plot_comparison
will run each command only once, as long as the output files are still there. Huzzah!
But we still have to specify the names of all three rules, in the right order, to run this. That's annoying! Let's fix that next.