Chapter 6 - using wildcards to make rules more generic
Let's take another look at one of those sketch_genomes_ rules:
rule sketch_genomes_1:
input:
"genomes/GCF_000017325.1.fna.gz",
output:
"GCF_000017325.1.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} --name-from-first
"""
There's some redundancy in there - the accession GCF_000017325.1 shows up
twice. Can we do anything about that?
Yes, we can! We can use a snakemake feature called "wildcards", which will let us give snakemake a blank space to fill in automatically.
With wildcards, you signal to snakemake that a particular part of an
input or output filename is fair game for substitutions using { and }
surrounding the wildcard name. Let's create a wildcard named accession
and put it into the input and output blocks for the rule:
rule sketch_genomes_1:
input:
"genomes/{accession}.fna.gz",
output:
"{accession}.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} \
--name-from-first
"""
What this does is tell snakemake that whenever you want an output file
ending with .fna.gz.sig, you should look for a file with that prefix
(the text before .fna.gz.sig) in the genomes/ directory, ending in
.fna.gz, and if it exists, use that file as the input.
(Yes, there can be multiple wildcards in a rule! We'll show you that later!)
If you go through and use the wildcards in sketch_genomes_2 and
sketch_genomes_3, you'll notice that the rules end up looking identical.
And, as it turns out, you only need (and in fact can only have) one rule -
you can now collapse the three rules into one sketch_genome rule again.
Here's the full Snakefile:
rule sketch_genome:
input:
"genomes/{accession}.fna.gz",
output:
"{accession}.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} --name-from-first
"""
rule compare_genomes:
input:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
output:
"compare.mat"
shell: """
sourmash compare {input} -o {output}
"""
rule plot_comparison:
message: "compare all input genomes using sourmash"
input:
"compare.mat"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot {input}
"""
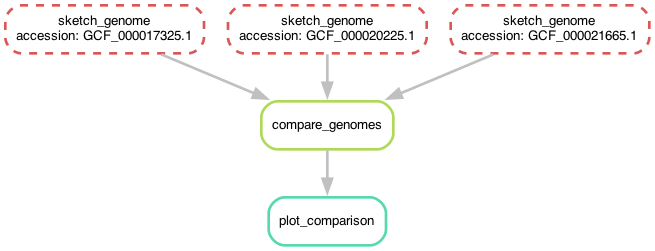
It looks a lot like the Snakefile we started with, with the crucial difference that we are now using wildcards.
Here, unlike the situation we were in at the end of last section where
we had one rule that sketched three genomes, we now have one rule
that sketches one genome at a time, but also can be run in parallel!
So snakemake -j 3 will still work! And it will continue to work as
you add more genomes in, and increase the number of jobs you want to
run at the same time.
Before we do that, let's take another look at the workflow now -
you'll notice that it's the same shape, but looks slightly different!
Now, instead of the three rules for sketching genomes having different names,
they all have the same name but have different values for the accession wildcard!